GSoC Project Summary and End-to-End Example
Google Summer of Code 2017 is approaching to its finish line. Time to sum up what has been completed! Check ModMashup.jl to get all the source code.
What?
The goal of the project was to replace GeneMANIA's network integration algorithm with a smaller memory footprint for high-performance computing. And, of course, having a command-line tool that can be integrated into any packages or system. The aim is to reduce the time needed to perform netDX query. Now we can say the project have successfully achieved this goal! (50x faster with promising accuracy)
How?
We design a novel method to replace linear regression part of GeneMANIA with a network embedding algorithm called Mashup, check Algorithm details here.
The main contribution of ModMashup.
Utilize network embedding to infer network weights.
Run cross validation in single query with a list of queries file, no more time is needed for re-initialization.
Only need similarity networks file and utilize julia's internal functionality to index patients' name to their id while GeneMANIA cost many time to construct Java database.
Quick Start
Required Dependencies
julia v0.5 +
You can download latest Julia from the official website. Version 0.5 or higher is highly recommended.
Installation
Enter Julia REPL.
$ juliaThen run the command below in Julia REPL.
Pkg.rm("ModMashup")
Pkg.clone("https://github.com/memoiry/ModMashup.jl")Example usage in Julia
Usage 1: Mashup Feature Selection
import ModMashup
cd(joinpath(Pkg.dir("ModMashup"), "test/data"))
#Set up database information
dir = "networks"
labels = "target.txt"
querys = "."
id = "ids.txt"
smooth = true
top_net = "nothing"
# Construct the dabase, which contains the preliminary file.
database = ModMashup.Database(dir, id,
querys, labels_file = labels,
smooth = smooth,
int_type = :selection,
top_net = top_net)
# Define the algorithm you want to use to integrate the networks
model = ModMashup.MashupIntegration()
# Running network integration
ModMashup.network_integration!(model, database)
net_weights = ModMashup.get_weights(model)
tally = ModMashup.get_tally(model)Usage 2: Mashup query runner for patients ranking using selected networks
import ModMashup
cd(joinpath(Pkg.dir("ModMashup"), "test/data"))
#Set up database information
dir = "networks"
querys = "CV_1.query"
id = "ids.txt"
smooth = true
# Top_networks contains selected top ranked networks.
top_net = "top_networks.txt"
# Construct the dabase, which contains the preliminary file.
database = ModMashup.Database(dir, id,
querys, smooth = smooth,
int_type = :ranking,
top_net = top_net)
# Define the algorithm you want to use to integrate the networks
int_model = ModMashup.MashupIntegration()
lp_model = ModMashup.LabelPropagation(verbose = true)
# Running network integration
ModMashup.fit!(int_model, lp_model, database)
# Pick up the result
#combined_network = ModMashup.get_combined_network(int_model)
net_weights = ModMashup.get_weights(int_model)
score = ModMashup.get_score(lp_model)
Mashup command tool
This project provide a Command Line Tool located in mashup.jl, which has two usage.
Modified Mashup feature selection.
Label propagation for patients ranking.
Arguments:
"command"
help = "what function do you want to use? ie. selection, ranking"
arg_type = String
required = true
"--net"
help = "Folder name where the similarity network is stored"
arg_type = String
required = true
"--id"
help = "Patients name in the database"
arg_type = String
required = true
"--labels"
help = "If for selection, it should be labels file name. If for ranking, it should be query file name and we use it to label patients."
arg_type = String
default = "nothing"
"--CV_query"
help = "If for selection, folder name where Query files stored. If for ranking, single query file name for use to label patients"
arg_type = String
"--top_net"
help = "This keyword is used for ranking, it should be file containing selected networks name."
arg_type = String
default = "nothing"
"--smooth"
help = "smooth the net or not"
arg_type = Bool
default = true
"--res_dir"
help = "where to put the result"
arg_type = String
"--cut_off"
help = "cut_off to select top ranked network in network integration"
arg_type = Int
default = 9Outputs:
For selection
networks_weights_with_name.txt: Txt file mapping networks name to its weights.mashup_tally.txt: Txt file mapping networks name to its tally.top_networks.txt: Txt file containing selected networks after cross validation.networks_index.txt: Txt file mapping networks name to its internal id.cv_query.txt: Txt file containing query internal id of each cross validation.beta.txt: Txt file containing beta vector.networks_weights_each_cv.txt: Txt file containing network weights of each cross validation.singular_value_sqrt.txt: Txt file containing sqrt of singular value.
For ranking
xxx_mashup_PRANK.txt: Txt file mapping patients name to their weights.xxx_mashup_NRANK.txt: Txt file mapping networks name to its weights.
Example
Usage 1: Mashup Feature Selection
First ensure that you have ModMashup.jl correctly installed in your computer.
$ var=$(julia -e "println(Pkg.dir())")
$ var="$var/ModMashup/test/data"
$ cd $var
$ mkdir temp_res
$ julia ../../tools/mashup.jl selection --net networks --id ids.txt --labels target.txt --CV_query . --smooth true --res_dir temp_resThe result will be saved at temp_res folder.
Usage 2: Mashup query runner for patients ranking using selected networks
After feature selection, you can run the command below to get patients ranking.
$ julia ../../tools/mashup.jl ranking --top_net temp_res/smooth_result/top_networks.txt --net networks --id ids.txt --CV_query CV_1.query --smooth true --res_dir temp_resThe result will be saved at temp_res folder.
End-to-End example in R
For those who want to use ModMashup in R and reproduce the experiment above.
You can use ModMashup command line tool for R's calling.
To wrap Julia's command line tool in R, I created two function to facilitate the procedure.One is
runMashup.R, which is the main function to call mashup command line tool. Another one ismashup_runCV_featureSet.R, a wrapper function aroundrunMashup.Rto facilitate selection of interested networks.
Experiment
Input
Mashup and GeneMANIA example shared same input.
TCGA Breast cancer dataset. Information used was patient ID and whether tumour is of subtype ‘Luminal A’ (LumA) or other.
N=348 patients with 232 as traning samples. Classes={LumA, other} annotation.
Similarity nets defined at the level of pathways, using Pearson correlation (ProfileToNetworkDriver) as similarity. Generates 1801 networks.
Result
Attention: Test needs to be repeated and the conclusion needs to be confirmed after changing the call to makePSN_NamedMatrix(), with writeProfiles=TRUE, I only test _con.txt file as currently ModMashup.jl only support that kind of format and so I have not make a experiment with .profile
This implementation in Julia is 50x faster(90 s ) than Java's implementation(4500 s) while achieving same and even better accuracy than raw GeneMANIA.
| Class | #total | #train | # selected networks | accuracy | PPV |
|---|---|---|---|---|---|
| LumA | 154 | 103 | 83 | 48/51(94.1%) | 48/58(81.4%) |
| other | 194 | 129 | 73 | 55/65(84.5%) | 55/58(94.9%) |
Table 1: netDX with ModMashup as kernel on BreastCancer dataset.
| Class | #total | #train | # selected networks | accuracy | PPV |
|---|---|---|---|---|---|
| LumA | 154 | 103 | 58 | 47/51(92.2%) | 47/58(81.0%) |
| other | 194 | 129 | 49 | 54/65(83.1%) | 54/58(93.1%) |
Table 2: netDX with GeneMANIA as kernel on BreastCancer dataset.
Relation between networks tally obtained from GeneMANIA and ModMashup
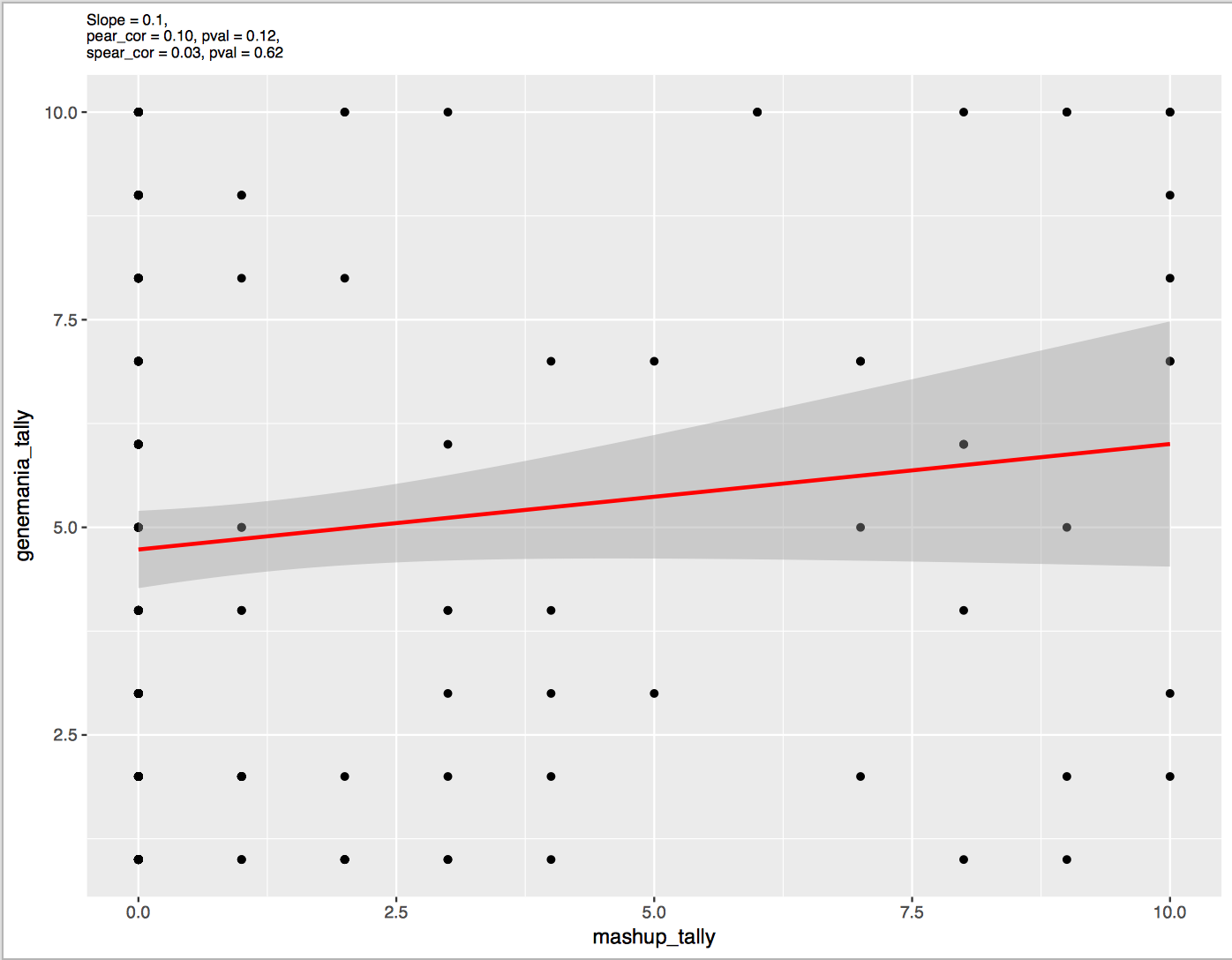
Figure 1: networks tally from GeneMANIA versus those from ModMashup for LumA type.
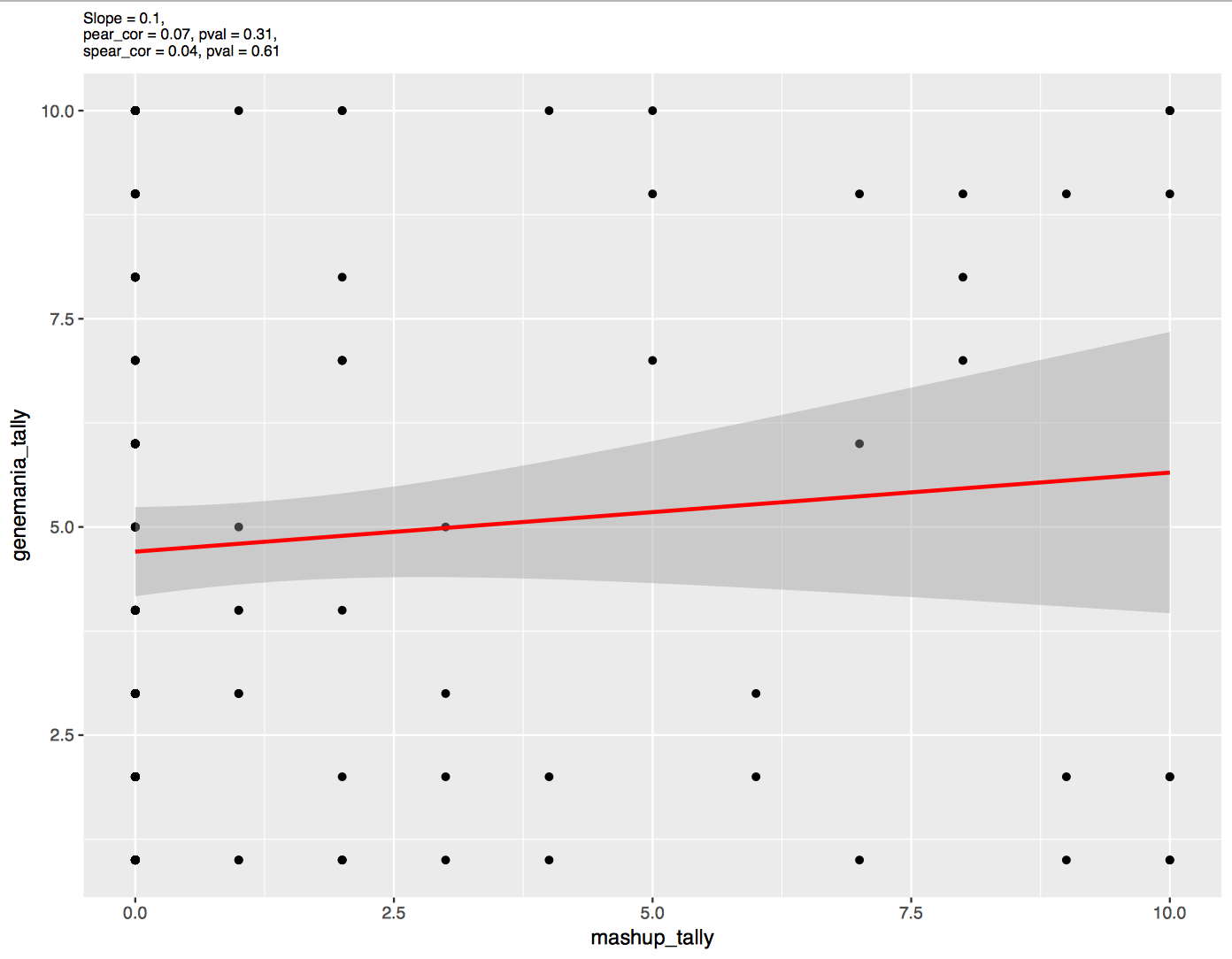
Figure 2: networks tally from GeneMANIA versus those from ModMashup for other type.
Relation between networks weight obtained from GeneMANIA and ModMashup
I have made two experiments to acquire network weight.
Correlation between H_cur and beta.
Dot product between H_cur and beta.
see GSoC report for more details about the experimental result.